
Fernando L. Dri
Multiscale Modeling of the Hierarchical Structure of Cellulose Nanocrystals
Abstract
A multiscale framework is being developed to predict and describe the thermo-mechanical behavior of cellulose nanocrystals using state-of-the-art computational tools capable of connecting atomistic based simulations to experiments through continuum based modeling techniques. As part of our efforts, we begin our investigation at the atomistic level using first-principle theories and molecular dynamics simulations to extract key mechanical properties (axial, bending and torsional stiffness) for isolated cellulose chains and single crystals as well as the thermal conductivity coefficients. A new continuum theory was developed to simulate cellulose nanocrystals using the finite element method.
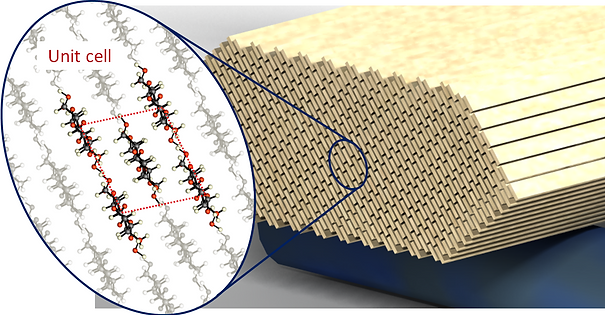
Figure 1: Schematic representation of a CNC modeled using continuum theories. At this level each cellulose chain is treated as a beam element, the system is linked together by non-bonded interactions (omitted for clarity)
Introduction
The goal of this work is to develop a multiscale framework that will allow us to generate a model capable of reproducing the behavior of cellulose nanocrystals (CNCs) and that can be extended to any material with similar characteristics such as other polysaccharides (e.g. α-chitin). To understand the internal behavior of CNC it is required to comprehend the role of bonded interactions (covalent bonds) and non-bonded interaction (van der Waals, Hydrogen bonds, Coulomb) in the mechanical and thermal properties as well as any other internal processes that can define the behavior of the entire system. The present work is based on the hypothesis that bonded and non-bonded interactions can be decoupled. Bonded interactions determine the cellulose chain behavior whereas non-bonded interactions define inter-chain behavior (i.e. size of the crystal and lattice parameters). The response of the crystal as a whole arises from the mutual work of these two mechanisms.
The computational approach employed uses a multiscale strategy to model the behavior of individual CNCs and CNC-based materials, connecting first principle calculations of CNCs to continuum models for CNC-based composite materials. Figure 2 depicts the summary of tools and approaches that we are currently following for this study. At the nanoscale, we perform explicit atomistic simulations to test our initial hypothesis and to progress in our path towards a complete understanding of the physic involved in the behavior of CNCs. Ab-initio modeling techniques like Quantum Mechanics – Density Functional Theory (QM-DFT) or Quantum Chemistry can be used to model atom-by-atom interactions in the most comprehensive way available today. While QM-DFT allows us to simulate hundreds of atoms in a picosecond timescale, molecular dynamics (MD) is capable of simulating times up to microsecond with millions of atom involved. This is achieved by reducing the details involved in each atomic interaction. With today’s computational power it is possible to simulate the behavior of an entire CNC crystal using MD.
However, the interaction between large CNCs and the behavior of the crystal as reinforcement material are still out of the reach of molecular dynamics simulation. Coarse-grained continuum mesoscale theories emerge as a possible solution to study the fundamental mechanical interaction of larger systems. Continuum models do not consider atomic interactions in the same way QM and MD do. Instead, the entire system (e.g., individual molecular chains or entire CNCs) can be treated as a whole and not as a group of individual atoms. Increasing the size and time length of the system is achieved by reducing the details involved in each length scale. For instance, going from MD to a continuum representation of cellulose chains, the results will highly depend on the force field parameterization being used to run the simulations. Keeping in mind those limitations, continuum theories have the advantage of simulating large systems that are far beyond the reach of MD. The key is to be able to capture the right physics at each length and time scale to feed the next scale with the right parameters. In our approach, we are no trying to fit variables to match simulations and experiments. The main goal is to connect several length scales accounting for the necessary mechanics and physics involved at each different scale to capture the right macroscopic behavior that ultimately can be compared with experiments.
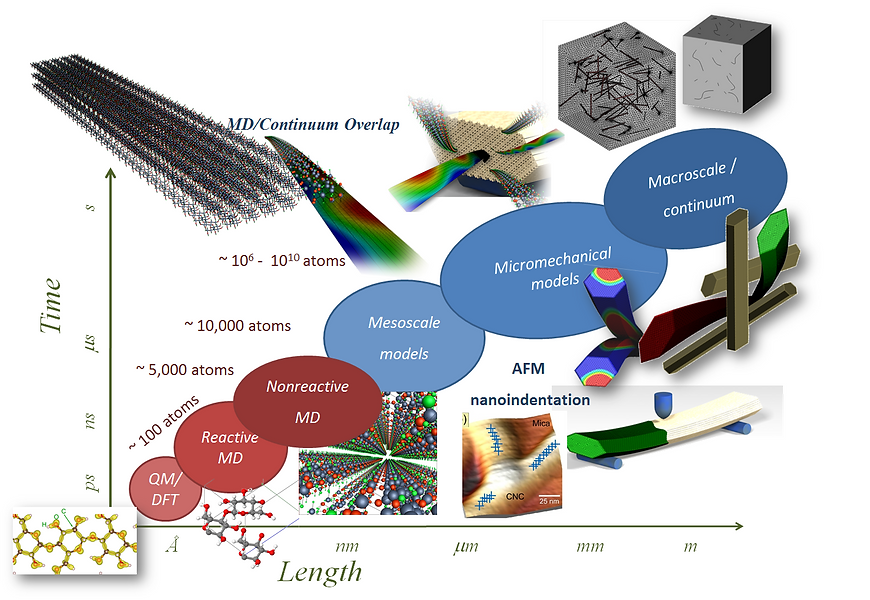
Figure 2: Time-length scale map. In red techniques that take into account atoms in an explicit way, in blue “continuum” theories.
Results
Our preliminary study using molecular dynamics demonstrated that simple continuum-based beam theories could indeed be used to simulate cellulose chains in a finite element framework. However, the non-bonded interaction needed additional development. As such, we developed special hydrogen bonds and wan der Waals interaction elements that can be integrated with the beam elements that represent the individual CNC chains. Figure 3 shows some details of the hydrogen-bond elements. In order to characterize and validate these new non-bonded theories, thought experiments borrowed from fracture mechanics were used to set up several MD simulations. A modified version of LAMMPS coupled with ReaFF force field was used in all the simulations discussed in this work. Two main groups of simulations were conducted, the first set consist in single chains of different lengths were the second involves crystals of several sizes.
All the previous results were used to construct a continuum model. Single chain simulations provided four key stiffness parameters that were used to feed our continuum beam model for cellulose chains. Single crystal molecular dynamics and quantum mechanics simulations provided insides of non-bonded interaction (van der Waals, Hydrogen bonds, Coulomb) that allowed us to fine tweak our hydrogen bond element. Overall, the two continuum elements working together allow us to fully model a cellulose nanocrystal using continuum theories.
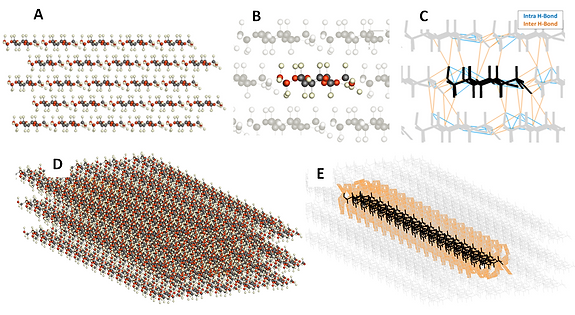
Figure 3: (A) 2D atomistic representation of a CNC. (B) Isolation of single cellulose chain inside the crystal. (C) Schematic representation of all possible H-bond interactions for a given chain. (D) 3D atomistic representation of a CNC. (E) Schematic representation of Inter-chain H-bonds of an interior chain. The number of H-bonds formed depends on the Force Field parameterization, figures only for illustration purposes.
Conclusions
A comprehensive solution to model cellulose nanocrystals from a continuum point of view was presented. Continuous theories can be applied to molecular systems under certain conditions but not without special care. The next step in our work is to simulate three point bending and nano-indentation experiments to correlate our models with experimental data. For more information on our current work see:
F.L. Dri, S. Shang, L.G. Hector Jr. Z-K. Liu, R. Moon, P.D. Zavattieri, "Extreme anisotropies of elasticity and thermal expansion in native cellulose Ib probed by Van der Waals density functionals", in preparation 2013.
F. Dri, L.G. Hector Jr. R. Moon, P.D. Zavattieri, "Quantum mechanics evaluation of three-dimensional elastic constant of cellulose Ib ", in preparation, 2013.